1. Definisi
Bioavailabilitas (ketersediaan hayati) didefinisikan sebagai kecepatan dan jumlah (rate and extent) bahan aktif atau senyawa aktif terserap dari produk obat dan menjadi tersedia di tempat kerjanya (FDA, 2014a). Karena ketersediaan di tempat kerja (site of action) seringkali berkaitan erat dengan kadar obat dalam darah, maka Pedoman ASEAN mengembangkan definisi tersebut menjadi kecepatan dan jumlah bahan aktif atau senyawa aktif terserap dari produk obat dan menjadi tersedia dalam peredaran darah (ASEAN, 2004). BPOM memberikan definisi yang dikaitkan dengan cara pengukuran bahan aktifnya, yakni persentase dan kecepatan zat aktif dalam suatu produk obat yang mencapai/tersedia dalam sirkulasi sistemik dalam bentuk utuh/aktif setelah pemberian produk obat tersebut, diukur dari kadarnya dalam darah terhadap waktu atau dari ekskresinya dalam urin (BPOM, 2005a). Definisi yang diberikan BPOM tersebut kurang sesuai untuk produk obat yang tidak ditujukan untuk diserap ke dalam sistem peredaran darah, misalnya obat yang ditujukan untuk obat yang bekerja lokal di saluran cerna.
Bioekivalensi (kesetaraan hayati) adalah tidak adanya perbedaan signifikan dalam kecepatan dan jumlah bahan aktif atau senyawa aktif dari produk ekivalen farmasetik atau alternatif farmasetik yang tersedia di tempat kerja obat jika diberikan pada dosis molar yang sama di bawah kondisi yang sama dalam penelitian yang didisain dengan tepat (FDA, 2014a) → (21 CFR 320.1 (e)). Dua produk obat disebut bioekivalen jika keduanya mempunyai ekivalensi farmasetik atau merupakan alternatif farmasetik dan pada pemberian dengan dosis molar yang sama akan menghasilkan bioavailabilitas yang sebanding sehingga efeknya akan sama, dalam hal efikasi maupun keamanan.(BPOM, 2005a; ASEAN, 2004; EMA, 2010)
Perbedaan antara bioavailabilitas dan bioekivalensi ada pada tujuan penelitian. Uji bioavalabilitas digunakan untuk menilai farmakokinetik dan kinerja produk obat terkait dengan penyerapan, distribusi, dan eliminasi obat in vivo. Sedangkan uji bioekivalensi menitikberatkan pada perbandingan formulasi berdasarkan analisa yang lebih difokuskan pada pelepasan bahan aktif (atau senyawa aktif) dari produk obat dan penyerapannya ke dalam peredaran sistemik.
Produk obat dapat dinyatakan sebagai ekivalen farmasetik jika mengandung bahan aktif yang identik, baik secara jenis maupun kekuatan, dalam bentuk sediaan dan jalur penghantaran yang sama. Produk ekivalen farmasetik juga harus memenuhi persyaratan kompendial atau standar lain yang berlaku, yakni dalam kekuatan, kualitas, kemurnian, dan identitas. Tetapi, produk ekivalen farmasetik tidak perlu sama dalam hal karakteristik seperti bentuk, konfigurasi garis pemecah (scoring), mekanisme pelepasan, kemasan, eksipien (termasuk pewarna, perisa, pengawet), waktu daluarsa, dan, dalam batas tertentu, penandaan (misalnya, ada atau tidaknya informasi farmakokinetik spesifik), dan cara penyimpanan (FDA, 2015). Adanya perbedaan eksipien dan/atau proses manufaktur dapat menyebabkan perbedaan kecepatan disolusi dan/atau penyerapan obat, sehingga produk yang ekivalen farmasetik tidak serta merta bioekivalen. Karena itu, perlu adanya pembuktian bahwa suatu produk yang ekivalen farmasetik juga bioekivalen untuk menjamin produk tersebut ekivalen terapetik
Produk obat dapat disebut alternatif farmasetik jika mengandung senyawa aktif (active moiety) yang sama, tetapi berbeda garam, ester, atau kompleks dari senyawa tersebut, atau berbeda bentuk sediaan atau kekuatan (contoh, tetrasiklin hidroklorida 250 mg kapsul dan tetrasikin fosfat kompleks 250 mg kapsul; kuinidin sulfat 200 mg tablet dan kuinidin sulfat 200 mg kapsul). Perbedaan bentuk sediaan dan kekuatan dalam satu line produk dari manufakturer yang sama dapat disebut sebagai alternatif farmasetik. Demikian halnya dengan produk lepas-lambat jika dibandingkan dengan produk lepas-segera dengan bahan aktif yang sama (Orange Book, 2015).
2. Penerapan uji bioavalabilitas dan bioekivalensi
Untuk obat baru dan produk obat baru, uji BA difokuskan pada penentuan bagaimana obat dilepas dari sediaan dan bergerak ke tempat kerjanya. (FDA, 2003a). Dokumentasi uji BA dapat digunakan untuk menilai kinerja produk obat yang digunakan dalam uji klinis untuk mendapatkan bukti keamanan dan efikasinya.
Dari sisi farmakokinetik, data uji BA digunakan untuk estimasi fraksi obat yang dapat diserap dari produk obat yang diberikan secara oral. Dari data tersebut dapat ditentukan fraksi relatif, jika dibandingkan dengan data BA sediaan larutan, suspensi, atau intravena (21 CFR 320.25(d)(2) dan (3)). Uji BA juga memberikan informasi farmakokinetik obat terkait dengan distribusi, eliminasi, efek nutrient terhadap penyerapan, proporsionalitas dosis, lienaritas farmakokinetik senyawa aktif dan, jika perlu, senyawa tidak aktif. Secara tidak langsung, data BA dapat memberikan informasi sifat obat sebelum masuk ke sirkulasi sistemik, seperti permeabilitas dan pengaruh enzim-enzim presistemik dan/atau transporter (misalnya, p-glikoprotein (FDA, 2003a).
Untuk obat baru dan produk obat baru, uji BE digunakan untuk membandingkan (1) formulasi awal dan akhir uji klinis; (2) formulasi yang digunakan dalam uji klinis dan uji stabilitasi, jika ada perbedaan; (3) formulasi uji klinis dan produk obat yang akan dipasarkan, jika ada perbedaan; (4) ekivalensi produk antar-potensi (FDA, 2014a). Sedangkan untuk obat copy baru, uji BE akan diminta pada saat registrasi untuk menunjukkan bahwa produk yang diregistrasikan ekivalen farmasetik dan bioekivalen terhadap produk referensi, yakni produk originator (FDA, 2003a).
Pasca persetujuan registrasi, uji BE juga bisa diterapkan jika terjadi perubahan pada formulasi dan/atau proses manufaktur, sepanjang daur hidup produk. FDA memberikan pedoman tingkat perubahan untuk menentukan apakah perubahan tersebut mengharuskan uji BE ulang atau cukup dengan uji komparasi in vitro. Pedoman FDA tersebut antara lain:
- SUPAC-IR (1995): Immediate Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation.
- SUPAC-MR (1997a): Modified Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation.
- SUPAC-SS (1997b): Nonsterile Semisolid Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Release Testing and In Vivo Bioequivalence Documentation.
Selain ketiga Pedoman SUPAC tersebut, terdapat satu dokumen tambahan sebagai pelengkap, yaitu SUPAC: Manufacturing Equipment Addendum (2014), untuk menentukan dokumentasi yang diperlukan dalam registrasi berdasarkan perubahan mesin produksi.
Uji bioekivalen tidak diperlukan untuk kasus tertentu, antara lain:
- Produk diberikan secara parenteral (intravena, subkutan, atau intramuskular) berupa larutan dalam air yang mengandung zat aktif yang sama, dalam kadar molar yang sama dengan produk komparator dan menggunakan eksipien yang sama atau mirip dalam kadar sebanding dengan yang ada dalam produk komparator. Eksipien tertentu (misalnya dapar, pengawet, dan antioksidan) bisa berbeda, tetapi harus ditunjukkan bahwa penggantian eksipien ini tidak berpengaruh terhadap keamanan dan/atau efikasi produk obat tersebut (BPOM, 2015). WHO (2015) menambahkan, hal yang sama berlaku juga untuk larutan berminyak parenteral. Namun, untuk produk ini, perlu dipastikan pemakaian pembawa minyak yang sama dengan yang dipakai dalam produk komparator. Demikian pula dengan larutan miselar, larutan yang mengandung bahan pengompleks, atau larutan yang mengandung pelarut campuran (co-solvent), harus mengandung eksipien fungsional yang sama secara jenis dan jumlahnya untuk menghindari uji ekivalensi. Penggantian eksipien fungsional tersebut dapat memicu pengkajian yang ketat;
- Produk ekivalen-farmasetik berupa larutan untuk pemakaian oral (misalnya sirup, eliksir, dan tinktur), yang mengandung zat aktif dalam kadar molar yang sama dengan produk komparator, mengandung eksipien fungsional yang sama dalam kadar yang sama (jika zat aktif tergolong BCS Kelas 1) dan eksipien yang sama dalam kadar yang sama (untuk zat aktif dari kelas BCS yang lain) (WHO, 2015). Terkait eksipien, BPOM (2015) menekankan bahwa eksipien yang digunakan hanya yang diketahui tidak mempunyai efek terhadap transit atau permeabilitas dalam saluran cerna, sehingga tidak mempengaruhi absorpsi atau stabilitas zat aktif selama berada dalam saluran cerna;
- Produk ekivalen-farmasetik berupa serbuk untuk rekonstitusi sebagai larutan dalam air dan hasil larutannya memenuhi kedua kriteria di atas;
- Produk ekivalen-farmasetik berupa gas;
- Produk ekivalen-farmasetik yang merupakan sediaan obat mata atau telinga, berupa larutan dalam air dan mengandung zat aktif yang sama dalam kadar molar yang sama dan mengandung eksipien yang sama dalam kadar yang sama. Eksipien tertentu (misalnya pengawet, dapar, bahan pengatur tonisitas atau kekentalan) bisa berbeda selama pemakaiannya tidak memberikan pengaruh terhadap bioavailabilitas, keamanan, dan/atau efikasi produk;
- Produk ekivalen-farmasetik yang merupakan sediaan topikal yang berupa larutan dalam air dan mengandung zat aktif yang sama dalam kadar molar yang sama dan mengandung eksipien yang sama dalam kadar yang sama. WHO (2015) memberikan catatan bahwa waiver tidak dapat diberikan pada bentuk sediaan topikal seperti gel, emulsi, atau suspensi, tetapi dapat diterapkan pada larutan berminyak jika komposisi pembawanya cukup sama;
- Produk ekivalen-farmasetik berupa larutan dalam air untuk nebulisasi atau tetes hidung, yang dapat digunakan dengan alat yang sama (atau tanpa alat), mengandung zat aktif yang sama dalam kadar yang sama dan mengandung eksipien yang sama dalam kadar yang sama. Produk dapat mengandung eksipien yang berbeda asalkan pemakaiannya tidak mempengaruhi bioavailabilitas, keamanan, dan/atau efikasi produk. WHO (2015) memberikan catatan bahwa waiver tidak dapat diberikan ke bentuk sediaan seperti suspensi untuk nebulisasi, tetes hidung dengan zat aktif berupa suspensi, semprot hidung dalam larutan atau suspensi, inhaler serbuk kering, atau inhaler dosis-terukur bertekanan berupa larutan atau suspensi.
Dalam kondisi (b), (c), (e), (f), dan (g) di atas pendaftar produk diwajibkan menunjukkan bahwa eksipien yang digunakan sama dan dalam kadar yang sama dengan yang ada dalam produk komparator, atau, seperti dalam kondisi (a), (e), dan (g), pemakaiannya tidak mempengaruhi bioavailabilitas, keamanan, dan/atau efikasi produk (WHO, 2015). Pembuktian pemakaian eksipien yang sama diwajibkan BPOM (2015) hanya untuk kondisi (e), (f), dan (g). Jika pendaftar tidak dapat memberikan informasi produk komparator dan BPOM juga tidak memiliki informasi tersebut, maka pendaftar diwajibkan untuk melakukan pengujian yang sesuai untuk menunjukkan perbedaan eksipien tidak mempengaruhi kinerja produk.
3. Metode uji bioekivalensi
Ada beberapa metode yang dapat digunakan dalam penentuan uji BE. Urutan berdasarkan prioritas dari pilihan utama hingga terakhir ditinjau dari tingkat akurasi, sensitivitas, dan reprodusibilitas, metode tersebut adalah:
- Uji farmakokinetik (PK)
- Uji farmakodinamik (PD)
- Uji klinis
- Uji in vitro
3.1. Uji farmakokinetik
Sejauh ini, uji perbandingan farmakokinetik merupakan pilihan utama dan paling banyak digunakan untuk produk obat yang diserap sistemik. Sedangkan uji perbandingan farmakodinamik dan klinis lebih umum digunakan untuk produk obat yang bekerja lokal.
3.2. Uji farmakodinamik
Uji PD tidak direkomendasikan selama obat diserap ke sirkulasi sistemik dan pendekatan PK dapat digunakan untuk penilaian BE. Hal ini disebabkan variabilitas pengukuran PD selalu lebih besar daripada PK. Selain itu, seringkali terjadi efek plasebo yang dapat makin memperburuk variabilitas dan memperumit disain eksperimen. Potensi munculnya efek plasebo harus dapat diantisipasi sebelumnya dalam disain penelitian dengan menambahkan fasa ketiga menggunakan plasebo dalam pengujian (WHO, 2006a).
Meskipun demikian, dalam kondisi pendekatan PK tidak dapat dilakukan, maka metode PD tervalidasi yang sesuai dapat digunakan untuk uji BE (FDA, 2003a). Kondisi khusus ini dapat ditemukan pada produk obat yang bekerja lokal dan beberapa produk obat yang bekerja sistemik tetapi kadarnya terlalu rendah untuk diukur dari cairan biologis atau adanya masalah keamanan jika digunakan pendekatan farmakokinetik untuk penilaian BE. Untuk produk yang bekerja lokal, alasan lain tidak menggunakan pendekatan PK adalah karena keberadaan obat dalam sirkulasi sistemik setelah pemberian produk tidak menggambarkan ketersediaan obat di lokasi kerjanya (FDA, 2003b).
Orlistat merupakan inhibitor selektif lipase dalam usus, yang digunakan dalam penanganan obesitas. Obat ini bekerja lokal dalam saluran cerna, sehingga tidak diperlukan penyerapan sistemik untuk efikasinya (Zhi et al, 1995). Karena efek hambatan yang dilakukan oleh orlistat, lipase tidak dapat menghidrolisis trigliserida dalam makanan menjadi asam lemak bebas dan monogliserida yang dapat diserap. Sebagai akibatnya, dapat terdeteksi terjadinya peningkatan ekskresi lemak dalam feses. Konsentrasi orlistat dalam plasma sporadik dan rendah (< 10 ng/mL atau 0,02 μM) sehingga tidak memadai untuk menggunakan pendekatan PK untuk uji BE (Zhi et al, 1999). Oleh karena itu, pendekatan PD melalui rasio jumlah eksreksi lemak feses selama 24 jam pada steady state terhadap jumlah lemak yang diberikan melalui makanan, digunakan dalam uji BE (FDA, 2010c).
3.3. Uji klinis
Jika pendekatan PK dan PD tidak memungkinkan, maka perbandingan uji klinis dapat dilakukan untuk penentuan BE. Namun, metode ini dinilai tidak sensitif sehingga sebaiknya dihindari jika ada pengganti yang lebih baik. Jumlah subyek yang sangat besar diperlukan untuk mendapatkan power statistik yang memadai. Sebagai contoh, diperlukan 8600 pasien untuk mendapatkan power statistik yang memadai untuk mendeteksi respon perbaikan 20% dari obat uji dibandingkan dengan plasebo. Contoh lain, diperlukan 2600 pasien infark miokardial untuk dapat menunjukkan penurunan resiko sebesar 16%. Perbandingan dua formulasi untuk uji BE berdasarkan titik akhir klinis akan memerlukan jumlah subyek yang lebih besar, tergantung pada variabilitas parameter sasaran dan rentang penerimaan (WHO, 2006a).
3.4. Uji in vitro
3.4.1. Biowaiver berdasarkan BCS
Uji in vitro jarang digunakan sendiri untuk uji BE. Namun, untuk saat ini, dengan penerapan BCS, biowaiver atau penggantian uji in vivo dengan uji in vitro, yakni uji profil disolusi terbanding, dapat dilakukan.
BCS (Biopharmaceutics Classification System) adalah pengelompokan bahan obat berdasarkan sifat kelarutan dalam air dan permeabilitas usus. Jika dikombinasikan dengan disolusi produk obat, BCS melibatkan tiga faktor yang dapat mempengaruhi BA suatu produk oral padat lepas segera, yakni disolusi, kelarutan, dan permeabilitas usus (Amidon et al, 1995). Berdasarkan BCS, bahan obat dapat dikelompokkan menjadi:
Kelas 1: Kelarutan tinggi – Permeabilitas tinggi
Kelas 2: Kelarutan rendah – Permeabilitas tinggi
Kelas 3: Kelarutan tinggi – Permeabilitas rendah
Kelas 4: Kelarutan rendah – Permeabilitas rendah
Definisi kelarutan tinggi dalam BCS berbeda dari definisi umum. Penentuan kelarutan tinggi dalam BCS didasarkan kemampuan melarut bahan obat dari produk obat dengan dosis tertinggi dalam ≤ 250 mL media air dalam rentang pH tertentu. Estimasi volume 250 mL tersebut diambil berdasarkan penerapan pemberian obat pada pasien puasa yang disertai dengan segelas air (sekitar 8 oz, kurang lebih setara dengan 250 mL), yang umum diterapkan dalam uji BE.
Pada awalnya, rentang yang digunakan adalah pH 1-8 (FDA, 1997c; EMA, 2001; ASEAN, 2004) atau 1- 7,5 (FDA, 2000). Pada 2006, WHO melakukan revisi definisi kelarutan tinggi, yakni jika dosis tertinggi obat dalam melarut dalam 250 mL atau kurang media air, dalam rentang pH 1,2-6,8 pada 37°C. Batas pH 6,8 ini menggambarkan bahwa obat harus terlarut sebelum mencapai bagian tengah jejunum, untuk memastikan penyerapannya dalam saluran cerna (WHO, 2006b). Revisi definisi ini selanjutnya diterapkan pula oleh EMA (2010) dan FDA (2015).
Kelarutan ditentukan dengan media dengan beberapa pH dalam rentang 1-6,8 pada suhu 37 ± 1°C. Kondisi pH yang digunakan dapat berdasarkan karakteristik ionisasi bahan obat, misalnya pada pH = pKa, pH = pKa + 1, pH = pKa – 1, pH 1, dan pH 6,8. EMA merekomendasikan sedikitnya dalam tiga media dapar, yakni pH 1,2; pH 4,5; dan pH 6,8; serta pada pH = pKa, selama masih dalam rentang pH 1,2-6,8. Penentuan kelarutan dalam masing-masing pH dilakukan minimal dengan 3 replikasi. Jika variabilitas saat pengujian tinggi, direkomendasikan memperbanyak jumlah replikasi untuk mendapatkan nilai kelarutan yang dapat dipercaya. Larutan dapar USP dapat digunakan, kecuali ada permasalahan secara fisik atau kimia dengan komponen obat. pH larutan di akhir pengujian perlu diukur kembali.
Suatu bahan obat dinyatakan memiliki permeabilitas tinggi jika jumlah terserap dalam saluran cerna mencapai ≥ 85% dari obat yang diberikan, berdasarkan penetapan keseimbangan massa (yang disertai pembuktian stabilitas dalam saluran cerna), atau dibandingkan pada BA absolut (FDA, 2015). Penggunaan batas 85% merupakan pelonggaran dari batas sebelumnya, 90%. Dengan adanya penyesuaian ini, beberapa obat yang sebelumnya masuk dalam BCS Kelas 3 berpindah ke BCS Kelas 1, misalnya parasetamol, aspirin, allopurinol, lamivudine, dan prometazin (WHO, 2006b).
Pembuktian stabilitas perlu digarisbawahi karena diperlukan untuk pembuktian bahwa kehilangan sejumlah obat dalam saluran cerna disebabkan oleh rendahnya permeabilitas atau karena mengalami degradasi. Uji stabilitas dalam saluran cerna dapat dilakukan dalam media lambung dan usus buatan. Larutan obat dalam media tersebut diinkubasi pada suhu 37°C selama jangka waktu tertentu, misalnya 1 jam dalam cairan lambung dan 3 jam dalam cairan usus. Penurunan kadar signifikan, yakni lebih dari 5%, dalam pengujian ini merupakan indikasi obat tidak stabil (FDA, 2015).
Kelas permeabilitas dapat ditentukan berdasarkan uji PK pada subyek manusia menggunakan metode keseimbangan massa dan BA absolut atau pendekatan perfusi usus. Selain itu, dapat juga dengan pengujian tanpa subyek manusia, misalnya perfusi usus in vivo atau in situ dengan model hewan, dan/atau metode permeabilitas in vitro menggunakan potongan jaringan usus, atau lapis tunggal sel epitel yang sesuai. Dari semua metode tersebut, yang paling diinginkan adalah data manusia. Pengujian yang lain hanya bersifat pendukung.
Permeabilitas prodrug tergantung pada mekanisme dan lokasi perubahan prodrug tersebut menjadi bahan obat. Jika pengubahan terjadi setelah penyerapan, maka permeabilitas prodrug tersebut yang perlu ditentukan. Sebaliknya, jika pengubahan terjadi sebelum penyerapan, maka yang ditentukan adalah permeabilitas dari bahan obat, sebagai hasil pengubahan.
FDA memberikan biowaiver untuk produk sediaan solida lepas segera, dengan bahan obat BCS kelas 1 dan kelas 3, yang menunjukkan disolusi in vitro cepat atau sangat cepat, dengan metode uji yang direkomendasikan, dan selama tidak ada bahan tambahan yang sediaan tersebut yang dapat mempengaruhi secara signifikan penyerapan bahan aktif (FDA, 2015).
Secara rinci, pengajuan biowaiver menurut FDA untuk produk obat BCS kelas 1 harus memenuhi persyaratan sebagai berikut:
- Bahan obat memiliki kelarutan tinggi dan permeabilitas tinggi
- Produk obat (uji dan referensi) cepat melarut, dan
- Produk obat tidak mengandung eksipien yang dapat mempengaruhi kecepatan dan jumlah penyerapan obat.
Untuk produk obat BCS kelas 3, FDA mempersyaratkan:
- Bahan obat memiliki kelarutan tinggi
- Produk obat (uji dan referensi) sangat cepat melarut, dan
- Formulasi produk uji sama secara jenis dan sangat mirip secara jumlah, yakni masuk dalam kategori perubahan tingkat 1 dan 2 SUPAC-IR, dari komposisi produk referensi.
Definisi cepat melarut (rapidly dissolving) adalah jika 85% atau lebih dari bobot label bahan aktif terlarut dalam waktu 30 menit dari produk obat lepas-segera , menggunakan metode keranjang 100 rpm atau metode dayung 50 rpm (atau 75 rpm, dengan justifikasi), dalam 500 mL atau kurang media: (1) HCl 0,1 N atau Cairan Lambung Buatan tanpa enzim; (2) dapar pH 4,5; dan (3) dapar pH 6,8 atau Cairan Usus Buatan tanpa enzim. Sedangkan, produk lepas-segera dinyatakan sangat cepat melarut (very rapidly dissolving) jika dalam kondisi disolusi yang sama, 85% atau lebih dari bobot label bahan aktif terlarut dalam waktu 15 menit.
Biowaiver berdasarkan BCS menurut EMA dapat diterapkan pada produk obat BCS kelas 1 dan kelas 3 (EMA, 2010), dengan persyaratan yang berbeda. Untuk produk obat BCS kelas 1 lepas segera dipersyaratkan:
- Bahan obat telah terbukti menunjukkan kelarutan tinggi dan penyerapan lengkap.
- Karakteristik disolusi in vitro produk uji dan produk referensi sangat cepat (>85% dalam 15 menit) atau cepat (85% dalam 30 menit), dalam metode uji yang direkomendasikan, dan
- Eksipien yang dapat mempengaruhi bioavailabilitas sama secara jenis dan jumlahnya. Secara umum, penggunakan eksipien yang sama dan dalam jumlah yang sama lebih diharapkan.
Sedangkan untuk produk obat BCS kelas 3 lepas segera, biowaiver berdasarkan EMA dapat diterapkan jika:
- Bahan obat telah terbukti menunjukkan kelarutan tinggi dan penyerapan terbatas.
- Karakteristik disolusi in vitro produk uji dan produk referensi sangat cepat (>85% dalam 15 menit) dalam metode uji yang direkomendasikan, dan
- Eksipien yang dapat mempengaruhi bioavailabilitas sama secara jenis dan jumlahnya, dan eksipien lain secara jenis sama dan secara jumlah sangat mirip.
Batas perbedaan relatif jumlah eksipien dari kedua produk obat oral padat untuk dapat disebut secara kuantitas sama diberikan dalam pedoman WHO (2015), sebagai berikut:
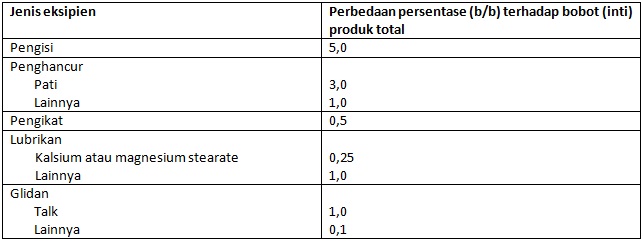
WHO (2006a) sempat memberikan biowaiver pada kelas BCS yang lebih luas, selain untuk produk obat BCS kelas 1 dan kelas 3, juga mencakup sebagian produk obat BCS kelas 2, yakni bahan aktif memiliki kelarutan tinggi pada pH 6,8 tetapi kurang melarut pada pH 1,2 dan pH 4,5 dan dengan permeabilitas tinggi. Namun, WHO (2015) kemudian menghilangkan biowaiver untuk produk obat BCS kelas 2 tersebut.
Sebaliknya, BPOM (2015) justru menerima biowaiver produk BCS kelas 2 dengan ketentuan zat aktif terdisolusi cepat pada pH 6,8 dan memiliki profil yang mirip dengan obat komparator pada pH yang lain. Juga berlaku jika disolusi kurang dari 10% pada salah satu pH.
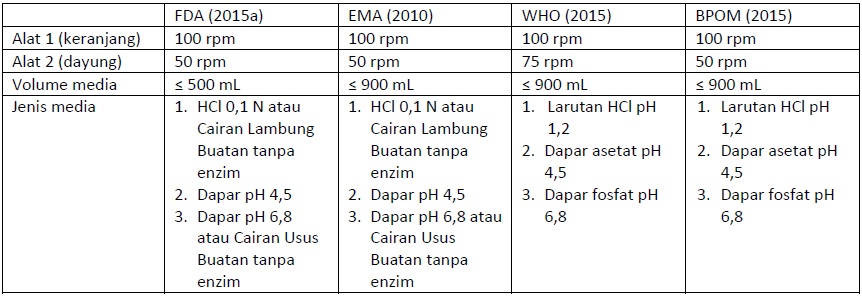
Metode uji disolusi yang direkomendasikan EMA hampir sama dengan FDA, kecuali pada kecepatan dayung, biasanya 50 rpm, dan volume media disolusi 900 mL atau kurang. Sedangkan, WHO menerapkan kecepatan untuk dayung 75 rpm dan volume media disolusi 900 mL atau kurang (Tabel 1).
Tabel 1 Perbandingan Kondisi Uji Disolusi yang Direkomendasikan FDA, EMA, dan WHO
Biowaiver berdasarkan BCS tidak dapat diterapkan untuk obat dengan indeks terapi sempit (contohnya, digoxin, fenitoin, teofilin, dan warfarin) atau obat yang ditujukan untuk diserap di rongga mulut (tablet sublingual, bukal) (FDA, 2015). BPOM (2015) menambahkan biowaiver tidak berlaku untuk produk obat dengan eksipien yang dapat mempengaruhi penyerapan zat aktif, seperti sorbitol, manitol, natrium lauril sulfat, dan surfaktan lainnya.
3.4.2. Pengaruh eksipien
Salah satu persyaratan biowaiver berdasarkan BCS adalah eksipien yang diharapkan sama dengan yang digunakan oleh produk komparator, dari jenis dan, dalam beberapa kasus, mencakup jumlahnya. Eksipien tersebut dapat mempengaruhi bioavailabilitas obat melalui efeknya terhadap:
- Motilitas saluran cerna
- Interaksi dengan bahan aktif, misalnya kompleksasi
- Permeabilitas obat
- Interaksi dengan transporter membrane (EMA, 2010)
Contoh eksipien yang telah diketahui memiliki pengaruh terhadap penyerapan obat adalah pemanis seperti sorbitol, dan manitol (Fassihi et al, 1991; Atkin et al, 1995). Eksipien tersebut tidak diserap dengan baik dari saluran cerna, tetapi dapat meningkatkan tekanan osmotik dalam usus. Sebagai akibatnya, terjadi peningkatan kecepatan pengosongan lambung dan memperpendek waktu transit usus. Jumlah obat yang dapat terserap tergantung pada kecepatan penyerapan dan lama obat berada di saluran cerna, khususnya usus. Perubahan waktu transit dalam saluran cerna dapat berpengaruh pada jumlah obat yang terserap, terutama obat yang memiliki permeabilitas rendah (Uppoor et al, 2014).
Chen et al (2007) melakukan penelitian efek dua macam gula, yaitu sukrosa dan sorbitol, terhadap bioavailabilitas ranitidin (obat dengan permeabilitas rendah) dan metoprolol (obat dengan permeabilitas tinggi). Sebagai hasilnya, terjadi penurunan signifikan Cmax dan AUC, masing-masing sekitar 50% dan 45%, ranitidin yang diberikan bersama sorbitol, dibandingkan dengan yang menggunakan sukrosa. Pada metoprolol, sorbitol menurunkan Cmax sekitar 23% tetapi tidak memberikan efek signifikan pada AUC. Chen et al (2013) juga menunjukkan bahwa manitol memiliki efek yang sama seperti sorbitol dan menurunkan bioavailabilitas simetidine, yang juga obat dengan permeabilitas rendah.
PEG 400 mempengaruhi penyerapan ranitidin dari saluran cerna (Basit et al, 2001). Pada kadar rendah, PEG 400 meningkatkan penyerapan, yang diduga disebabkan pengubahan permeabilitas ranitidin di usus, sedangkan pada kadar tinggi, PEG 400 justru menurunkan penyerapan yang diduga disebabkan penurunan waktu transit usus (Basit et al, 2002; Schulze et al, 2003). Peningkatan penyerapan yang disebabkan PEG 400 dilaporkan spesifik terhadap gender tertentu, yakni peningkatan BA ranitidine terjadi hanya pada subyek pria, sedangkan pada subyek wanita tidak terpengaruh (Ashiru et al, 2008).
PEG dengan bobot molekul rata-rata 400-20000 juga diketahui menjadi contoh eksipien yang dapat berinteraksi dengan pompa efluks P-glikoprotein intestinal (Shen et al, 2006). Transporter P-gp ini dapat mengembalikan sebagian obat yang memasuki sel membran dinding usus balik ke lumen sehingga menurunkan jumlah obat terabsorpsi. PEG dapat masuk ke sel dan berinteraksi dengan P-gp. Polimer dengan bobot molekul rendah (rata-rata 750-2000) melintasi membran sel secara difusi pasif, sedangkan yang memiliki bobot molekul tinggi (rata-rata 5000-20000) masuk ke sel, dalam konsentrasi rendah, secara difusi pasif, dan dalam konsentrasi tinggi, secara endositosis yang diperantarai kaveolae (Wang et al, 2020). Absorpsi obat yang merupakan substrat P-gp dapat terpengaruh karena penggunaan bahan tambahan yang dapat mengubah kinerja pompa efluks ini. Dalam kasus perbedaan formulasi terkait penggunaan eksipien yang mempengaruhi absorpsi, profil disolusi obat uji bisa sangat mirip dengan obat komparator tetapi profil farmakokinetik berbeda.
Bahan lain yang dapat memberikan efek pada BA obat adalah surfaktan, seperti polysorbate 80 dan SLS. Garcia-Arieta (2014) memberikan contoh bahwa perbedaan SLS 2 mg saja sudah cukup memberikan perbedaan BA obat BCS kelas 2.
3.4.3. Biowaiver berdasarkan potensi
Biowaiver berdasarkan potensi dapat diberikan jika (1) produk obat dalam bentuk sediaan yang sama, tetapi dalam potensi yang berbeda; (2) perbedaan potensi ini memiliki komposisi bahan aktif dan tambahan yang secara proposional sama terhadap produk yang dengan potensi yang digunakan dalam uji BE; dan (3) produk dengan potensi yang baru memenuhi uji disolusi in vitro yang sesuai (FDA, 2003). EMA (2010) dan WHO (2015) menambahkan satu persyaratan yang harus dipenuhi untuk mendapatkan biowaiver tersebut, yakni produk dibuat dengan proses manufaktur yang sama. BPOM (2015) mempersyaratkan juga obat dengan kekuatan yang berbeda dibuat oleh produsen yang sama di tempat produksi yang sama.
Definisi ‘sama secara proposional’ berdasarkan FDA (2003):
- Semua bahan aktif dan tambahan benar-benar dalam proporsi yang sama pada potensi yang berbeda (contoh, tablet dengan potensi 50 mg memiliki semua bahan aktif dan tambahan setengah dari tablet dengan potensi 100 mg, dan dua kali dari tablet dengan potensi 25 mg).
- Bahan aktif dan bahan tambahan tidak dalam proporsi yang benar-benar sama antarpotensi, tetapi rasio bahan tambahan terhadap bobot total unit sediaan berada dalam batas perubahan tingkat 1 dan tingkat 2 SUPAC-IR dan SUPAC-MR. Definisi ini tidak disebutkan dalam pedoman FDA 2014a.
- Untuk bahan aktif yang poten, dengan bobot bahan aktif per unit sediaan rendah, bobot total per bentuk sediaan hampir sama pada semua potensi (dalam ± 10% dari bobot total dari potensi yang digunakan dalam uji BE), bahan tambahan yang sama digunakan pada semua potensi, dan perubahan potensi hanya diperoleh dari penyesuaian sejumlah yang diperlukan dari bahan aktif dan satu atau lebih bahan tambahan. Perubahan persentase bahan tambahan berada dalam batas perubahan tingkat 1 dan tingkat 2 SUPAC-IR dan SUPAC-MR.
Dalam pedoman yang lebih baru, FDA (2014a) juga menjelaskan defisini ‘sama secara proporsional’ untuk tablet bilayer. Tablet dua lapis tersebut dipertimbangkan sebagai satu formulasi meski terdiri dari dua lapisan terpisah dengan komposisi berbeda. Untuk menilai kesamaan proporsional dari potensi tablet yang berbeda, semua komponen dari kedua lapisan tersebut harus sama secara proporsional. Jika satu lapis sama secara proporsional, sedangkan satu lapis lainnya tidak, maka secara keseluruhan tablet tersebut tidak sama secara proporsional. Hal ini wajar karena adanya kemungkinan perbedaan interaksi antara lapisan dari tablet antarpotensi, sebagai akibat adanya perbedaan ukuran lapisan dan jumlah eksipien yang ada pada tiap lapisan.
Definisi ‘sama secara proporsional’ menurut EMA (2010) adalah rasio antara jumlah tiap bahan tambahan terhadap jumlah bahan aktif sama untuk semua potensi (untuk produk lepas segera, komponen penyalut, cangkang kapsul, pewarna, dan perisa tidak perlu mengikuti aturan ini). Kondisi yang masih dapat dikategorikan sama secara proporsional, untuk bobot bahan aktif kurang dari 5% dari bobot tablet inti atau bobot isi kapsul: (1) bobot bahan tambahan tablet inti atau isi kapsul sama dengan sediaan yang digunakan untuk uji BE, hanya bobot bahan aktifnya yang berubah, atau (2) bobot bahan pengisi berubah, menyesuaikan perubahan jumlah bahan aktif. Jumlah bahan tambahan lain dalam tablet inti atau isi kapsul tetap sama. Definisi ini diadopsi oleh ASEAN (2004).
WHO (2015) memberikan definisi ‘sama secara proporsional’ yang hampir sama dengan FDA:
- Semua bahan aktif dan tambahan benar-benar dalam proporsi yang sama pada potensi yang berbeda (contoh, tablet dengan potensi 50 mg memiliki semua bahan aktif dan tambahan setengah dari tablet dengan potensi 100 mg, dan dua kali dari tablet dengan potensi 25 mg). Untuk produk lepas segera, komponen penyalut, cangkang kapsul, bahan pewarna, dan perisa tidak diharuskan mengikuti aturan tersebut.
- Untuk bahan aktif yang poten, dengan bobot bahan aktif per unit sediaan rendah (hingga 10 mg per unit sediaan atau tidak lebih dari 5% bobot sediaan), bobot total per bentuk sediaan hampir sama pada semua potensi (dalam ± 10% dari bobot total), bahan tambahan yang sama digunakan pada semua potensi dengan hanya bobot zat aktif yang berubah, atau jumlah pengisi yang berubah sesuai perubahan bobot zat aktif, sedangkan jumlah eksipien yang lain pada semua kekuatan sediaan.
Biowaiver berdasarkan potensi tidak bisa diberikan jika obat menunjukkan kinetika eliminasi yang tidak linear (FDA, 2003a). Sebagai contoh, klaritromisin yang memiliki klirens yang lebih rendah pada sediaan dengan potensi yang lebih tinggi karena metabolisme di hati yang dapat jenuh (Davey, 1991; Chu et al, 1993). Karena perbedaan ini kinetika ini, maka kedua potensi, 500 mg dan 250 mg, harus diuji BE (FDA, 2010a).
EMA (2010) dan WHO (2015) tidak merekomendasikan pemakaian surfaktan dalam pengambilan profil disolusi. BPOM (2015) masih mengizinkan penggunaan surfaktan hingga 1%. Untuk kekuatan yang berbeda, jika produk uji menunjukkan perbedaan profil disolusi karena masalah kondisi tunak (sink condition), pengujian dapat dilakukan dengan menggunakan dosis yang sama per bejana, misalnya dua tablet 5 mg terhadap satu tablet 10 mg.
Untuk tablet lepas tunda, jika kekuatan yang berbeda memiliki formulasi yang secara proporsional sama dengan produk yang diuji BE, kekuatan yang lebih rendah dapat diberikan biowaiver jika menunjukkan profil disolusi yang sama, f2 tidak kurang dari 50, dalam media asam (pH 1,2) selama dua jam, dan diikuti dengan disolusi pada pH 6,8 (WHO, 2015; BPOM, 2015).
Evaluasi proporsi penyalut enterik atau lepas tunda bukan didasarkan pada penambahan bobot, melainkan pada luas permukaan, dengan penambahan bobot per luas permukaan tablet (mg/cm2) yang sama (WHO, 2015).
Untuk tablet lepas lambat, jika kekuatan yang berbeda memiliki formulasi yang secara proporsional sama untuk zat aktif dan eksipiennya, dengan mekanisme pelepasan obat yang sama, uji BE dapat dilakukan hanya pada kekuatan tertinggi. Sedangkan, kekuatan yang lebih rendah dapat diberikan biowaiver jika menunjukkan profil disolusi yang sama dengan kekuatan tertinggi, dalam larutan dapar dengan 3 pH berbeda, antara pH 1,2 dan 7,5, dan dalam media QC sesuai prosedur uji yang direkomendasikan. WHO (2015) memberikan contoh titik waktu pengambilan sampel uji disolusi untuk produk lepas lambat 12 jam: 1, 2, 4, 6, 8, dan 12 jam; dan untuk produk lepas lambat 24 jam: 1, 2, 4, 6, 8, 16, dan 24 jam.
Untuk tablet lepas lambat dengan mekanisme pelepasan pompa osmotik, uji profil disolusi terbanding cukup dilakukan dalam kondisi uji yang direkomendasikan (WHO, 2015).
Untuk kapsul, yang tiap kekuatan mengandung pelet lepas tunda atau lepas lambat yang sama, hanya berbeda jumlah peletnya, uji profil disolusi terbanding (f2 ≥ 50) dalabm kondisi uji yang direkomendasikan sudah mencukupi untuk biowaiver berdasarkan proporsi dosis formulasi.
4. Pengaruh makanan
Uji BA efek makanan umumnya dilakukan terhadap obat atau produk obat baru, untuk menilai pengaruh makanan terhadap kecepatan dan jumlah obat yang diserap jika obat diberikan dalam waktu segera setelah makan, jika dibandingkan dengan kondisi tanpa makanan. Sedangkan uji BE efek makanan dilakukan untuk obat copy, untuk melihat efek makanan terhadap produk dibandingkan dengan obat referensi (FDA, 2002). FDA memberikan biowaiver untuk produk obat copy jika produk uji dan produk referensi cepat melarut, memiliki profil disolusi yang sama, dan mengandung obat BCS kelas 1.
Makanan dapat mengubah BA obat dan mempengaruhi BE antara produk uji dan produk referensi. Pengaruh makanan ini dapat disebabkan melalui beberapa cara:
- Penundaan waktu pengosongan lambung
- Stimulasi aliran empedu
- Pengubahanan pH gastrointestinal
- Peningkatan aliran darah splanchnic
- Pengubahan metabolisme luminal bahan obat
- Interaksi fisika atau kimia dengan bahan obat atau produk obat.
Kandungan nutrisi dan kalori makanan, volume makanan, dan suhu makanan dapat memyebabkan perubahan fisiologis saluran cerna, yang berpengaruh pada perubahan waktu transit produk obat, disolusi luminal, permeabilitas obat, dan ketersediaan sistemik. Secara umum, makanan dengan total kalori dan kandungan lemak yang tinggi memberikan pengaruh paling besar terhadap BA obat. Oleh karena itu, untuk uji efek makanan terhadap BA dan BE, FDA merekomendasikan penggunaan makanan kalori tinggi (sekitar 800-1000 kalori) dan lemak tinggi (sekitar 50 persen dari kandungan kalori total dari makanan). Jika diuraikan lebih lanjut, makanan yang digunakan dalam pengujian tersebut mengandung sekitar 150 kalori protein, 250 kalori karbohidrat, dan 500-600 kalori lemak (FDA, 2002).
4.1. Penundaan waktu pengosongan lambung
Banyak faktor yang mempengaruhi waktu pengosongan lambung, antara lain volume isi lambung, pH, hasil pencernaan makanan berkalori (lemak, protein, karbohidrat) dan kalori total, osmolaritas, viskositas, dan suhu isi lambung.
Produk obat BCS kelas 1 memiliki kelarutan yang tinggi dan tidak tergantung pada pH. Untuk formulasi cepat melarut produk obat BCS kelas 1, efek makanan lebih disebabkan oleh penundaan waktu pengosongan lambung, terutama pada Cmax dan Tmax obat tersebut.
Penundaan waktu pengosongan lambung dapat menyebabkan peningkatan BA obat yang memiliki kelarutan buruk dalam air, karena adanya lebih banyak waktu bagi obat untuk melarut sebelum tiba di lokasi penyerapannya. Sebagai contoh, BA nitrofurantoin meningkat karena adanya makanan yang menyebabkan penundaan waktu pengosongan lambung sehingga disolusi meningkat. Oleh karena itu, penggunaan nitrofurantoin direkomendasikan bersama dengan makanan untuk meningkatkan penyerapan obat.
Sebaliknya, untuk obat yang tidak stabil, lamanya waktu dalam lingkungan yang tidak bersahabat di lambung justru dapat menurunkan BA. Contoh obat yang tidak stabil dalam kondisi asam adalah didanosin. Pemberian bersama makanan dapat menurunkan Cmax dan AUC hingga 55% dibandingkan dengan kondisi puasa. Dengan demikian, pemakaian didanosin direkomendasikan dalam kondisi perut kosong.
Pengeluaran produk dari lambung juga dapat dipengaruhi oleh karakteristik obat sendiri. Produk obat yang tidak terdisintegrasi di lambung, misalnya tablet salut enterik, dapat tertahan di lambung dan keluar dalam waktu yang bervariasi sehingga memberikan hasil yang tidak menentu (EMA, 2014).
Makanan memberikan efek kecil pada waktu transit usus. Dalam kondisi dengan atau tanpa makanan, waktu transit usus sekitar 4 jam. Faktor yang lebih berpengaruh terhadap penyerapan dan BA obat yang menjadi pembeda antara kondisi dengan atau tanpa makanan di usus adalah perubahan pH, viskositas, aktivitas enzimatik, kompleksasi, khelasi, dan halangan fisik yang dapat terjadi dalam lumen usus halus (DeHaven dan Conner, 2014).
4.2. Stimulasi aliran empedu
Empedu akan dilepas dari kantung empedu ke duodenum setelah makan. Fungsi empedu dalam proses pencernaan makanan adalah sebagai surfaktan yang membantu pembentukan emulsi dari lemak makanan sehingga meningkatkan penyerapan lemak tersebut. Dengan adanya makanan yang mengandung lemak tinggi, obat yang sukar larut dan bersifat lipofilik mengalami peningkatan kelarutan dan disolusi karena adanya kenaikan garam empedu, dibandingkan kondisi tanpa makanan (Charman et al, 1997). Akibatnya, obat dengan sifat ini dapat mengalami peningkatan BA.
Sebaliknya, kenaikan garam empedu yang dipicu makanan berlemak tinggi ini justru berpotensi menurunkan kelarutan dan disolusi obat hidrofilik, sehingga mengalami penurunan BA. Atenolol merupakan contoh senyawa hidrofilik yang dapat mengalami perubahan BA berupa penurunan 30% AUC dan 28% Cmax karena pengaruh empedu (Barnwell et al, 1993).
4.3. Perubahan pH gastrointestinal
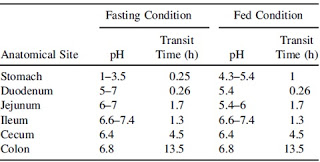
Perbedaan pH (dan waktu transit) masing-masing bagian saluran cerna dapat dilihat pada Tabel 2. Nilai pH tersebut dapat bervariasi antarindividu dan dapat dipengaruhi oleh banyak faktor, seperti usia, aktivitas fisik, dan kondisi kesehatannya.
Saat ada makanan yang masuk, terjadi peningkatan sekresi asam dalam lambung. Namun, jumlah asam lambung yang meningkat tersebut tidak dapat menyebabkan penurunan pH, justru terjadi sebaliknya. Hal ini dapat terjadi karena kemampuan makanan untuk mendapar atau mengencerkan asam yang ada dalam cairan lambung. Peningkatan pH hingga sekitar pH 5 dapat terjadi sekitar 10 menit setelah pertama kali makanan masuk, dan kembali ke pH kondisi puasa sekitar 1,5-2 jam kemudian.
Perubahan pH ini tentu akan berpotensi mempengaruhi kelarutan dan BA bahan obat. Senyawa basa lemah umumnya lebih mudah larut dalam kondisi asam. Peningkatan pH dalam lambung bisa jadi dapat menyebabkan penurunan kelarutan dan disolusi obat yang bersifat basa lemah. Bahkan, dalam kondisi waktu pengosongan lambung yang lama dan adanya efek makanan, justru terjadi pengendapan. Ketoconazol merupakan contoh obat basa lemah yang mengalami penurunan BA akibat kenaikan pH lambung (Charman et al, 1997).
Hal yang sebaliknya dapat terjadi pada obat asam lemah. Peningkatan pH lambung dapat menyebabkan peningkatan kelarutan dan disolusi, sehingga terjadi peningkatan penyerapan obat.
Produk obat yang diformulasi sebagai produk lepas-termodifikasi, seringkali menggunakan mekanisme pelepasan yang tergantung pH. Misalnya, produk salut enterik yang didisain untuk tidak melepas obat dalam kondisi asam, dan mulai melepas obatnya setelah mencapai lingkungan dengan pH yang lebih tinggi di usus. Jika polimer yang digunakan melarut pada pH 5, maka peningkatan pH lambung setelah makan berpotensi menyebabkan pelepasan obat yang lebih dini dari seharusnya. Jika menggunakan polimer yang baru melarut pada pH yang lebih tinggi, misalnya pH 6, justru pada kondisi setelah makan dapat terjadi penundaan pelepasan obat karena pH dalam usus harus bagian awal (duodenum dan jejunum) belum mencapai pH yang dapat memecah polimer penyalut tersebut.
4.4. Peningkatan aliran darah splanchnic
Peningkatan aliran darah splanchnic dapat mempengaruhi penyerapan obat, terutama yang melalui mekanisme difusi pasif (penyerapan paraseluler). Dalam kondisi aliran darah splanchnic rendah, kadar obat lintas membran menjadi cepat setimbang. Sebaliknya, saat terjadi peningkatan aliran darah splanchnic, kesetimbangan kadar obat lintas membran menjadi sulit tercapai sehingga obat dapat terdifusi dengan lebih cepat dan meningkatkan BA.
Dari segi metabolisme prasistemik, peningkatan aliran darah splanchnic yang meningkatkan penyerapan pasif, juga menyebabkan peningkatan efek lintas pertama terhadap obat. Sebagai akibatnya, peningkatan metabolisme ini dapat memberikan penurunan terhadap efek positif peningkatan aliran darah splanchnic terhadap BA.
Obat yang diserap melalui mekanisme selain difusi pasif tidak dipengaruhi secara signifikan oleh peningkatan aliran darah splanchnic.

Bagian-bagian saluran cerna (Wen dan Park, 2010)
pH dan Waktu Transit Tiap Bagian Saluran Cerna (Wen dan Park, 2010)
4.5. Pengubahan metabolisme luminal bahan obat
BA obat dikendalikan oleh proses penyerapan dan pembersihannya dari sistem sirkulasi (systemic clearance). Banyak contoh obat yang diserap dengan sangat baik, tetapi menunjukkan BA yang rendah akibat transformasi metabolik prasistemik di mukosa usus dan/atau efek lintas pertama di hati. Labelatol adalah contoh obat yang banyak mengalami metabolisme prasistemik sehingga memiliki BA yang rendah. Pemakaian obat ini bersama makanan dilaporkan dapat menaikkan BA 38%, sebagai akibat penurunan klirens prasistemik tersebut (Daneshmend dan Roberts, 1982). Metabolisme hepatic dan mukosa usus seringkali bersifat dapat-jenuh, sehingga dapat dipengaruhi oleh kecepatan penyerapan obat.
Contoh makanan, atau lebih tepatnya minuman, yang paling sering disebut dapat mempengaruhi klirens prasistemik adalah jus grapefruit (sejenis jeruk, bukan anggur). Jus grapefruit telah menunjukkan dapat meningkatkan BA obat seperti calcium channel blocker, benzodiazepin, dan imunosupresan. Mekanisme utama interaksi ini adalah hambatan pada CYP3A, enzim metabolism utama di usus, oleh furanokumarin yang terkandung dalam jus. Jus grapefruit juga menghambat transporter efluks obat, P-glycoprotein. Transporter ini memompa obat yang sudah terserap kembali ke arah lumen usus. Efek hambatan pada keduanya berakibat pada kenaikan BA, namun seringkali jadi bermasalah akibat munculnya efek samping obat karena tingginya kadar obat dalam plasma (Yamada et al, 2014).
4.6. Interaksi fisika atau kimia obat-makanan
Efek makanan terhadap penyerapan obat dapat berupa interaksi langsung antara obat dengan sesuatu di dalam makanan. Misalnya, serat makanan bisa mengikat obat sehingga menyebabkan penurunan BA obat tersebut.
Pembentukan khelat dapat terjadi antara dengan logam yang ada dalam makanan dengan obat. Siprofloksasin dapat membentuk khelat dengan kation multivalensi, seperti aluminum, besi, magnesium, dan kalsium, dalam saluran cerna sehingga terjadi penurunan BA. Neuhofel et al (2002) melaporan penurunan AUC dan Cmax siprofloksasin hingga 21% dan 22% jika diberikan bersama dengan jus jeruk yang difortifikasi kalsium, dibandingkan dengan jus jeruk tanpa fortifikasi.
Keberadaan makanan juga dapat menjadi penghalang fisik obat untuk bisa mencapai membran lokasi penyerapan. Proses pencernaan menghasilkan medium kental yang secara signifikan menurunkan kecepatan difusi obat dalam cairan lumen (Fleisher et al, 1999).
4.7. Kondisi pemakaian khusus – penaburan (sprinkler)
Beberapa produk obat, misalnya kapsul berisi butiran pelet lepas terkendali, dapat direkomendasikan digunakan dengan menaburkan di atas makanan lunak dan ditelan tanpa dikunyah. Untuk produk obat baru, jika cara pemakaian obat dapat dilakukan dengan cara demikian dan disebutkan pada brosur, maka perlu dilakukan uji BA tambahan, yaitu dengan cara penaburan, dan kemudian dibandingkan dengan produk yang langsung ditelan utuh. Kedua pengujian dilakukan dalam kondisi perut kosong. Untuk obat copy baru, FDA juga meminta uji BE tambahan dengan prosedur penaburan (FDA, 2002).
Untuk produk obat dengan butiran pelet yang ditujukan untuk ditaburkan, ukuran butiran maksimal 2,8 mm (10% variasi dari target ukuran 2,5 mm). Ketentuan ukuran ini ditujukan untuk menghindari refleks mengunyah, yang biasa muncul jika dalam mulut terasa butiran yang besar (FDA, 2012a).
5. Disain studi BE
Untuk perbandingan dua formulasi, disain yang direkomendasikan adalah randomized, 2-periods, 2-sequences, cross over design. Antar-perlakukan harus dipisahkan oleh periode wash-out, yang memastikan bahwa kadar obat berada di bawah limit deteksi pada semua subyek, sebelum dilakukan periode kedua. Umumnya, untuk mencapai kadar obat hingga di bawah kemampuan deteksi alat itu diperlukan waktu 5 kali waktu paruh atau lebih (EMA, 2010). Periode wash-out minimal tujuh hari (WHO, 2006a).
Sebagai alternatif, untuk kondisi tertentu dapat digunakan, yakni (1) disain paralel, untuk obat dengan waktu paruh sangat panjang; dan (2) disain replikat, untuk obat dengan variasi PK yang tinggi. Disain replikat crossover dapat digunakan untuk estimasi variabilitas intrasubyek (within-subject) dari produk uji dan/atau produk referensi, menggunakan replikasi parsial (3-periode) atau penuh (4-periode). Jumlah sampel yang diperlukan untuk replikasi parsial lebih banyak daripada replikasi penuh untuk bisa mendapatkan power statistik yang setara untuk penetapan BE (FDA, 2001). Dalam hal jumlah subyek, disain replikat memiliki keuntungan memerlukan jumlah subyek yang lebih kecil dibandingkan disain 2 periode untuk mendapatkan power statistik yang sama (FDA. 2003a).
Contoh penerapan disain replikat dalam pedoman FDA adalah uji BE untuk progesteron (FDA, 2011b) dan warfarin natrium (FDA, 2012c).
5.1. Obat dengan waktu paruh panjang
Sebagaimana disebutkan di atas, obat dengan waktu paruh panjang (≥24 jam), dapat digunakan disain paralel sebagia alternatif. Tetapi, jika disain crossover lebih dikehendaki untuk digunakan, selama obat menunjukkan variabilitas intrasubyek rendah dalam distribusi dan klirens, maka AUC yang dipotong pada 72 jam (AUC0-72 jam) dapat digunakan sebagai pengganti AUC0-t atau AUC0-∞. Sebaliknya, untuk obat yang menunjukkan variabilitas intrasubyek yang tinggi dalam distribusi dan/atau klirens, maka penerapan AUC0-72 jam tidak diperbolehkan (FDA, 2003a).
5.2. Uji dosis tunggal vs dosis ganda
FDA lebih merekomendasikan uji PK dosis tunggal (single-dose) dibandingkan uji dosis ganda (multiple-dose), baik untuk produk obat lepas-segera maupun lepas-termodifikasi dalam rangka uji BE karena umumnya lebih sensitif untuk digunakan dalam menilai pelepasan bahan obat dari produk obat ke sirkulasi sistemik (FDA, 2003a).
Meskipun demikian, dalam beberapa kasus uji dosis ganda atau uji keadaan tunak (steady state) dapat digunakan. Contohnya, karena masalah keamanan maka uji BE untuk klozapin tablet dilakukan terhadap pasien yang sedang dalam masa perawatan dengan menggunakan obat yang sama. Pasien tersebut sedang menerima dosis harian yang tetap dengan dosis yang sama dalam interval 12 jam. Pasien yang mendapatkan dosis lebih tinggi dari 100 mg dalam interval 12 jam juga masih dapat diikutkan dalam pengujian dengan menggunakan dosis yang sebelumnya sudah digunakan. Berdasarkan jadwal acak, pasien dalam jumlah yang sama menerima produk uji dan produk referensi, dalam dosis yang sama, sebelum pengujian setiap 12 jam selama 10 hari. Setelah itu, produk yang diberikan kepada pasien dalam ditukar untuk 10 hari periode kedua. Tidak ada periode washout di antara kedua periode tersebut. Setelah uji selesai, pasien dapat terus melanjutkan terapinya menggunakan produk klozapin yang diresepkan dokternya (FDA, 2005).
Situasi yang lain yang memungkinkan penerapan uji dosis ganda adalah:
- Obat menunjukkan kinetik non-liear pada kondisi tunak (misalnya, metabolisme yang dapat jenuh, sekresi aktif)
- Dalam kondisi sensitifitas pengujian sangat rendah untuk bisa memeriksa profil PK hanya dari dosis tunggal
- Bentuk sediaan lepas lambat dengan kecenderungan akumulasi (sebagai tambahan untuk uji dosis tunggal).
5.3. Subyek sehat vs pasien
Berdasarkan EMA (2010) dan FDA (2003a), pengujian BE umumnya diterapkan pada subyek sehat berumur 18 atau lebih. WHO (2006a), ASEAN (2004), dan BPOM (2005) memberikan batasan antara 18 dan 55 tahun. Secara khusus, jika obat digunakan untuk orang tua, FDA merekomendasikan penggunaan sebanyak mungkin subyek dengan usia 60 tahun ke atas. Hal ini dilakukan karena ada risiko perbedaan antara kedua populasi usia, seperti waktu pengosongan lambung, pH, dan/atau waktu transit saluran cerna, yang dapat berpengaruh pada BA bahan obat.
Berat badan subyek, berdasarkan Indeks Massa Tubuh, EMA mempersyaratkan antara 18,5-30. Sedangkan, ASEAN dan BPOM menggunakan syarat antara 18-25. Kriteria sehat diambil berdasarkan uji laboratorium klinis (hematologi rutin, fungsi hati, fungsi ginjal, gula darah, dan urinalisis), riwayat penyakit dan pemeriksaan fisik. Subyek tidak boleh memiliki sejarah penyalahgunaan obat atau alkohol, dan sebaiknya bukan perokok. Untuk perokok, ASEAN dan BPOM memberikan batasan yang jelas bahwa perokok sedang (hingga 10 batang per hari) masih dapat diikutkan, namun pengaruhnya terhadap uji perlu didiskusikan.
Jika obat digunakan untuk kedua jenis kelamin, maka kedua jenis kelamin tersebut harus dilibatkan secara seimbang dalam pengujian. Penggunaan hanya salah satu jenis kelamin dapat dilakukan jika produk obat hanya ditujukan penggunaannya untuk jenis kelamin tertentu atau dinilai berbahaya jika diterapkan pada jenis kelamin yang berlainan. [lupa baca di mana]
Penggunaan subyek sehat pada di banyak prosedur uji BE bertujuan untuk menurunkan bias karena kondisi subyek yang lebih terkontrol. Tetapi, karena alasan keamanan, penggunakan subyek sehat dalam uji BE produk tertentu dapat digantikan oleh pasien, dengan catatan pasien tersebut dalam kondisi stabil dari segi penyakit dan pengobatannya selama masa uji BE. Tergantung pada karakteristik obat, indikasi, profil keamanan dan/atau efikasi obat, pengujian dapat dilakukan menggunakan disain crossover dan/atau paralel. Salah satu contoh produk obat yang menggunakan pasien sebagai subyek sudah dibahas di atas, klozapin. Contoh lain adalah produk obat onkologi, misalnya everolimus tablet (FDA, 2012b).
5.4. Dosis uji
Jika ada beberapa potensi produk, maka yang perlu diuji BE hanya satu atau dua potensi. Potensi yang lain dapat diajukan untuk biowaiver selama komposisinya memenuhi persyaratan sama secara proporsional dengan komposisi yang diuji BE. Dosis yang biasa digunakan dalam uji BE adalah dosis tertinggi yang dipasarkan.
Berdasarkan WHO (2006a), dosis yang lebih tinggi, dengan menggunakan dua unit produk, dapat dilakukan jika ditemukan kesulitan masalah analisis. Namun, dalam kasus ini total dosis tidak boleh lebih dari dosis harian tertinggi. Sebagai alternatif, untuk mengatasi masalah terkait sensitivitas metode pengujian ini dapat juga dengan pemotongan AUC hingga 3 x median tmax produk referensi.
Penggunaan dosis yang lebih rendah dapat dilakukan karena alasan keamanan. Sebagai contoh, sekali lagi, klozapin. Produk tablet klozapin ada lima potensi, yakni 12,5; 25; 50; 100; dan 200 mg. Pedoman uji BE FDA menggunakan dosis 100 mg untuk uji BE (FDA, 2005).
Jika obat menunjukkan PK yang tidak linear antarpotensi, maka uji BE harus dilakukan pada produk dengan potensi tertinggi dan terendah, atau potensi dalam rentang linear. EMA (2010) juga menambahkan bahwa jika kondisi non-linear tersebut bukan disebabkan oleh keterbatasan kelarutan, melainkan kejenuhan transporter, maka uji BE dapat dilakukan dengan produk potensi terendah atau potensi dalam rentang linear.
6. Senyawa yang diuji
6.1. Obat induk vs metabolit
Senyawa yang diukur dari sampel cairan biologis dalam uji BA dan BE adalah bahan aktif obat atau senyawa aktifnya (parent drug) dan, jika memungkinkan, metabolit aktifnya (FDA, 2013a). Untuk uji BA, FDA merekomendasikan pengukuran baik senyawa induk maupun metabolit aktif utamanya, jika secara metode analisis memungkinkan. Sedangkan untuk uji BE, pengukuran umumnya hanya pada senyawa induk yang dilepas dari produk obat. Hal ini didasari alasan bahwa profil kadar-waktu senyawa induk lebih sensitif terhadap perbedaan formulasi dibandingkan metabolit. Pemeriksaan terhadap metabolit lebih menggambarkan pembentukan metabolit, distribusi, dan eliminasi (FDA, 2003a). Hal ini juga berlaku untuk prodrug yang inaktif, uji BE tetap dilakukan berdasarkan pengukuran senyawa induk, sedangkan metabolit aktif tidak perlu diukur. Tetapi, untuk prodrug yang memiliki kadar rendah dalam plasma dan cepat tereliminasi sehingga menyulitkan penentuan BE berdasarkan senyawa induk, maka penggunaan metabolit aktif saja dapat dilakukan. Perlu digaris-bawahi, bahwa ini berlaku hanya untuk prodrug yang inaktif, yang tidak atau hanya memiliki sedikit konstribusi terhadap efikasi (EMA, 2010).
FDA merekomendasikan pemeriksaan metabolit dalam uji BE jika metabolit tersebut memiliki kontribusi tinggi terhadap efikasi dan /atau keamanan. Sebagai contoh, sulfasalazine (SSZ) dan balsalazid yang merupakan obat untuk ulcerative colitis. SSZ mengandung mesalamin yang diikat sulfapiridin melalui ikatan azo. Sedangkan, pada balsalazid, mesalamin diikat, juga dengan ikatan azo, dengan 4-aminobenzoil-B alanin (4-ABA), yang bersifat inert. Pengikatan ini mencegah obat diserap hingga akhirnya sesampainya di kolon, ikatan tersebut dipecah menggunakan enzim azoreduktase yang diproduksi oleh bakteri kolon, sehingga melepas mesalamin yang merupakan senyawa aktif. Untuk uji BE produk SSZ, sulfapiridin direkomendasikan untuk juga diperiksa, selain SSZ dan mesalamin, terkait masalah efek samping SSZ yang lebih banyak disebabkan oleh senyawa ini (FDA, 2010d). Sedangkan dalam uji BE balsalazid, FDA hanya merekomendasikan pengukuran kadar balsalazid dan mesalamin. 4-ABA tidak perlu diperiksa karena secara farmakologi tidak aktif dan tidak ada masalah keamanan (FDA, 2013b).
6.2. Enansiomer vs rasemat
Pengukuran rasemat menggunakan metode achiral (non-stereoselective) secara umum dapat diterima oleh FDA, EMA, WHO, dan BPOM (FDA, 2013a; EMA, 2010; WHO, 2006a; BPOM, 2005). Pengukuran enansiomer individual direkomendasikan jika ditemukan semua kondisi berikut:
- Enansiomer menunjukkan perbedaan PK
- Enansiomer menunjukkan perbedaan PD
- AUC enansiomer berbeda akibat adanya perbedaan kecepatan penyerapan masing-masing enansiomer. FDA: adanya penyerapan nonlinear antara enansiomer.
- Efikasi dan keamanan utamanya ditentukan oleh enansiomer minor.
Jika satu enansiomer aktif secara farmakologi, sedangkan yang lainnya tidak atau hanya sedikit aktif, EMA merekomendasikan pemeriksaan cukup dilakukan untuk enansiomer yang aktif saja.
6.3. Campuran kompleks
Beberapa obat dapat mengandung lebih dari satu senyawa aktif. Dalam kasus ini, tidak perlu dilakukan pemeriksaan terhadap semua senyawa yang ada dalam obat tersebut, melainkan cukup menentukan marker, yang dapat menunjukkan jumlah dan kecepatan penyerapan. Meskipun berbeda untuk tiap kasus, pemilihan marker dilakukan berdasarkan kriteria jumlah senyawa tersebut dalam produk, kadar dalam darah atau plasma, dan aktivitas biologi relatif terhadap senyawa yang lain dalam campuran. Jika pendekatan PK tidak bisa dilakukan dalam penetapan jumlah dan kecepatan penyerapan, maka pendekatan in vitro dapat diterapkan. Jika cara in vitro ini pun tidak bisa dilakukan, maka dapat dipertimbangkan pendekatan PD atau uji klinis (FDA, 2003; BPOM 2005).
6.4. Senyawa endogen
Beberapa bahan aktif obat merupakan senyawa endogen, yang secara normal ada dalam tubuh, baik karena diproduksi oleh tubuh atau berasal dari makanan normal. Jika senyawa endogen tersebut identik dengan bahan obat, tentu saja ini menyebabkan masalah tersendiri karena bahan obat yang ditambahkan, yang akan ditentukan BE-nya, tidak dapat dibedakan dengan yang sudah ada dalam tubuh. Jika kadar senyawa endogen cukup stabil sebelum dan selama pengujian, maka dapat dilakukan koreksi garis-dasar dalam evaluasi BE. Dalam kasus ini, tingkat garis-dasar bisa ditentukan dari rata-rata data yang sampel yang diambil dari beberapa titik waktu sebelum pengujian dengan obat (time-averaged baseline). Namun, jika kadar dalam tubuh naik-turun secara periodik, maka tingkat garis-dasar masing-masing harus ditentukan sesuai setiap interval pengambilan sampel (time-mached baseline).
Senyawa endogen juga dapat memiliki proses homeostatik, sehingga pemberian senyawa eksogen dapat menurunkan produksi senyawa endogen dalam tubuh sehingga mempengaruhi kadar sistemiknya. Untuk menurunkan masalah terkait homeostatik ini dan juga dapat untuk menghilangkan kebutuhan koreksi garis-dasar, FDA (2014a) merekomendasikan penggunaan pasien dengan produksi senyawa endogen rendah atau bahkan tidak mampu memproduksi senyawa tersebut sebagai subyek uji BE, daripada penggunaan subyek sehat. Sebagai contoh, uji BE untuk estradiol menggunakan wanita post-menopausal, baik secara fisiologi maupun akibat pembedahan (FDA, 2010b). Pemilihan populasi ini, selain karena merupakan target pengobatan, juga karena garis-dasar yang relatif stabil.
Jika senyawa endogen dipengaruhi oleh makanan, maka perlu dilakukan kontrol makanan yang ketat sebelum dan selama uji BE berlangsung. Sebagai contoh, dalam pedoman uji BE untuk KCl, FDA merekomendasikan subyek diberi makanan terstandarisasi, dengan jumlah asupan kalium, natrium, kalori, dan cairan diketahui. Subyek juga ditempatkan dalam ruangan dengan kondisi lingkungan terkontrol. Aktivitas fisik dihindari untuk mencegah keringat berlebih yang dapat menyebabkan kehilangan kalium. Makanan kecil dan minum diberikan dalam waktu dan jumlah yang tertentu. (FDA, 2011a)
6.5. Produk obat kombinasi atau diberikan bersamaan
Dua atau lebih bahan aktif obat dapat diformulasi menjadi produk obat tunggal, yang disebut sebagai produk obat kombinasi. Secara umum, tujuan uji BA terhadap produk kombinasi adalah membandingkan jumlah dan kecepatan penyerapan setiap bahan aktif dalam produk kombinasi tersebut terhadap jumlah dan kecepatan penyerapan masing-masing bahan aktif yang diformulasikan sebagai produk tunggal.
Dalam kasus tertentu, produk obat dapat diberikan bersamaan dengan produk obat yang lain (bukan diformulasikan bersama dalam satu produk tunggal), dengan tujuan untuk meningkatkan paparan salah satu obat (subject drug). Obat kedua tidak ditujukan untuk memberikan efek terapi, tetapi diperlukan untuk meningkatkan paparan sistemik obat subyek. Jika uji BE dilakukan untuk produk obat subyek, maka pemberiannya juga harus dilakukan bersama dengan produk obat kedua, baik untuk produk uji maupun produk referensi. Penentuan BE hanya dilakukan berdasarkan PK obat subyek, sedangkan obat kedua tidak perlu diukur kadarnya. Jika produk obat kedua memerlukan uji BE, maka uji tersebut dilakukan hanya pada produk obat kedua, tanpa pemberian produk obat subyek (FDA, 2014a).
Pustaka
Adkin DA, Davis SS, Sparrow RA, Huckle PD, Phillips AJ, Wilding IR (1995) The effects of pharmaceutical excipients on small intestinal transit. B. J. Clin. Pharmac. 39:381-387
Amidon GL, Lennernäs H, Shah VP, Crison JR (1995) A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12(3): 413-420
ASEAN (2004) Guidelines for the Conduct of Bioavailability and Bioequivalence Studies. http://www.hsa.gov.sg/content/dam/HSA/HPRG/Western_Medicine/Overview_Framework_Policies/Guidelines_on_Drug_Registration/ACTR_GuidelinesforConductofBioavailabilityandBioequivalenceStudies_Nov05.pdf
Ashiru DA, Patel R, Basit AW (2008) Polyethylene glycol 400 enhances the bioavailability of a BCS class III drug (ranitidine) in male subjects but not females. Pharm. Res. 25(10):2327-2333
Barnwell SG, Laudanski T, Dwyer M, Story MJ, Guard P, Cole S, Attwood D (1993) Reduced bioavailability of atenolol in man: the role of bile acids. Int. J. Pharm. 89(3): 245-250
Basit AW, Newton JM, Short MD, Waddington WA, Ell PJ, Lacey LF (2001) The effect of polyethylene glycol 400 on gastrointestinal transit: implications for the formulation of poorly-water soluble drugs. Pharm. Res. 18(8):1146-1150
Basit AW, Podczeck F, Newton JM, Waddington WA, Ell PJ, Lacey LF (2002) Influence of polyethylene glycol 400 on the gastrointestinal absorption of ranitidine. Pharm. Res. 19(9):1368-1370
BPOM (2005) Peraturan Kepala Badan Pengawas Obat dan Makanan No HK.00.05.3.1818 Tahun 2005 tentang Pedoman Uji Bioekivalensi.
BPOM (2015) Pedoman Uji Bioekivalensi. Jakarta: Badan POM
Charman WN, Porter CJH, Mithani S, Dressman JB (1997) Physicochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J. Pharm. Sci. 86(3): 269-282
Chen ML, Straughn AB, Sadrieh N, Meyer M, Faustino PJ, Ciavarella AB, Meibohm B, Yates CR, Hussain AS (2007) A modern view of excipient effects on bioequivalence: case study of sorbitol. Pharm. Res. 24(1): 73-80
Chen ML, Sadrieh N, Yu L (2013) Impact of osmotically active excipients on bioavailability and bioequivalence of BCS class III drugs. AAPS J. 15(4):1043-1050.
Chen ML (2014) Fundamentals of Bioequivalence. Dalam: Yu LX dan Li BV (eds) FDA Bioequivalence Standards. New York: Springer, pp. 29-53
Chu S, Wilson DS, Deaton RL, Mackenthun AV, Eason CN, Cavanaugh JH (1993) Single- and multiple-dose pharmacokinetics of clarithromycin, a new macrolide antimicrobial. J. Clin. Pharmacol. 33(8): 719-726
Daneshmend TK, Roberts CJC (1982) The influence of food on the oral and intravenous pharmacokinetics of a high clearance drug: a study with labelatol. Br. J. Clin. Pharmacol.14:73-78
DeHaven WI dan Conner DP (2014) The Effects of Food on Drug Bioavailability and Bioequivalence. Dalam: Yu LX dan Li BV (eds) FDA Bioequivalence Standards. New York: Springer, pp. 95-118
Davey PG (1991) The pharmacokinetics of clarithromycin and its 14-OH metabolite. J. Hosp. Infect. 19 Suppl A: 29-37
EMA (2001) Note for Guidance on the Investigation of Bioavailability and Bioequivalence, London, 20 Juli 2001. CPMP/EWP/QWP/1401/98. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003519.pdf
EMA (2010) Guideline on the Investigation of Bioequivalence. London, 20 Januari 2010. CPMP/EWP/QWP/1401/98 Rev. 1/Corr http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
EMA (2014) Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms. EMA/CPMP/EWP/280/96 Corr 1 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/11/WC500177884.pdf
Fassihi AR, Dowse R, Robertson SSD (1991) Influence of sorbitol solution on the bioavailability of theophylline. Int. J. Pharm. 72( ):175-178
FDA (1995) Guidance for Industry: SUPAC-IR: Immediate Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070636.pdf
FDA (1997a) Guidance for Industry: SUPAC-MR: Modified Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070640.pdf
FDA (1997b) Guidance for Industry: SUPAC-SS: Nonsterile Semisolid Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Release Testing and In Vivo Bioequivalence Documentation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070930.pdf
FDA (1997c) Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070237.pdf
FDA (2000) Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System.
FDA (2001) Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070244.pdf
FDA (2002) Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070241.pdf
FDA (2003a) Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. http://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf
FDA (2003b) Guidance for Industry: Bioavailability and Bioequivalence Studies for Nasal Aerosols and Nasal Sprays for Local Action. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070111.pdf
FDA (2005) Draft Guidance on Clozapine. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM249219.pdf
FDA (2010a) Draft Guidance on Clarithromycin. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201285.pdf
FDA (2010a) Draft Guidance on Estradiol. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM238055.pdf
FDA (2010b) Draft Guidance on Orlistat Capsules. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201268.pdf
FDA (2010c) Draft Guidance on Sulfasalazine. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM199673.pdf
FDA (2011a) Draft Guidance on Potassium Chloride. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM270390.pdf
FDA (2011b) Draft Guidance on Progesterone. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM209294.pdf
FDA (2012a) Draft Guidance for Industry: Size of Beads in Drug Products Labeled for Sprinkle. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm240243.pdf
FDA (2012b) Draft Guidance on Everolimus. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM249239.pdf
FDA (2012c) Draft Guidance on Warfarin Sodium. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201283.pdf
FDA (2013a) Draft Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM377465.pdf
FDA (2013b) Draft Guidance on Balsalazide Disodium. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM358111.pdf
FDA (2014a) Draft Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs – General Considerations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM389370.pdf
FDA (2014b) Guidance for Industry: SUPAC: Manufacturing Equipment Addendum. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM346049.pdf
FDA (2015a) Draft Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf
FDA (2015b) Draft Guidance for Industry: Dissolution Testing and Specification Criteria for Immediate-Release Solid Oral Dosage Forms Containing Biopharmaceutics Classification System Class 1 and 3 Drugs. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM456594.pdf
Fleisher D, Li C, Zhou Y, Pao L, Karim A (1999) Drug, meal and formulation interactions influencing drug absorption after oral administration: clinical implications. Clin. Pharmacokinet. 36(3): 233-254
Garcia-Arieta A (2014) Interaction between active pharmaceutical ingredients and excipients affecting bioavailability: impact on bioequivalence. Eur. J. Pharm. Sci.65(18): 89-97
Neuhofel AL, Wilton JH, Victory JM, Hejmanowski LG, Amsden GW (2002) Lack of bioequivalence of ciprofloxacin when administered with calcium-fortified orange juice: a new twist on an old interaction. J. Clin. Pharmacol. 42(4): 461-466
Orange Book (2015) Approved Drug Products with Therapeutic Equivalence Evaluations, 35th Edition. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Office of Generic Drugs.
Schulze JD, Waddington WA, Eli PJ, Parsons GE, Coffin MD, Basit AW (2003) Concentration-dependent effects of polyethylene glycol 400 on gastrointestinal transit and drug absorption. Pharm. Res. 20(12): 1984-1988
Shen Q, Lin Y, Handa T, Doi M, Sugie M, Wakayama K, Okada N, Fujita T, Yamamoto A (2006) Modulation of intestinal P-glycoprotein function by polyethylene glycols and their derivatives by in vitro transport and in situ absorption studies. Int. J. Pharm. 313(1-2): 49-56
Uppoor RS, Vaidyanathan J, Mehta M, Yu LX (2014) Biowaiver and Biopharmaceutic Classification System. Dalam: Yu LX dan Li BV (eds) FDA Bioequivalence Standards. New York: Springer, pp. 119-137
Wang T, Guo Y, He Y, Ren T, Yin L, Fawcett JP, Guo J, Sun H (2020) Impact of molecular weight on the mechanism of cellular update of polyethylene glycols (PEGs) with particular reference to P-glycoprotein. Acta Pharm. Sin. B 10(10): 2002-2009
Wen H dan Park K (2010). Introduction and Overview of Oral Controlled Release Formulation Design. Dalam: Wen H dan Park K (Eds) Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice. New Jersey: John Wiley & Sons, pp. 1-19
WHO (2006a) Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability. Annex 7, WHO Technical Report Series, No. 937. https://apps.who.int/iris/bitstream/handle/10665/43443/WHO_TRS_937_eng.pdf
WHO (2006b) Proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release, solid oral dosage forms. Annex 8, WHO Technical Report Series, No. 937. https://apps.who.int/iris/bitstream/handle/10665/43443/WHO_TRS_937_eng.pdf
WHO (2015) Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability. Annex 7, WHO Technical Report Series, No. 992. https://www.who.int/medicines/areas/quality_safety/quality_assurance/expert_committee/WHO_TRS_992_web.pdf
Yamada S, Misaka S, Ito Y, Watanabe H, Umegaki K (2014) Effects of Natural Products on Pharmacokinetics and Pharmacodynamics of Drugs. Dalam: Folkerts G dan Garssen J (eds.) Pharma-Nutrition. New York: Springer, pp. 189-211
Zhi J, Melia AT, Eggers H, Joly R, Patel IH (1995) Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J. Clin. Pharmacol. 35(11):1103-1108
Zhi J, Mulligan TE, Hauptman JB (1999) Long-term systemic exposure of orlistat, a lipase inhibitor, and its metabolites in obese patients. J. Clin. Pharmacol.39(1):41-46
terimakasih kakkk, sangat menambah wawasan 😍 semangat dan sukses buat kedepannya kakk
ReplyDelete